■ 蛋白質轉譯修飾的新篇章
The New Chapter of Protein Modification Research
The New Chapter of Protein Modification Research

蛋白質轉譯修飾 Protein Modification
|
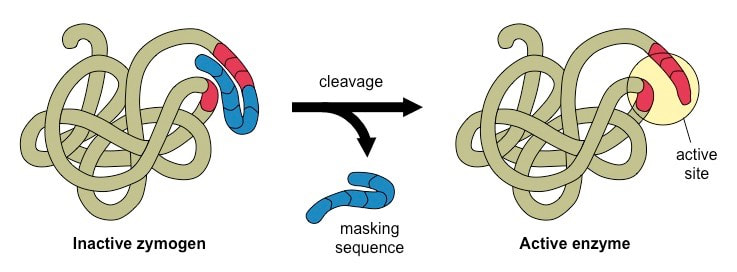
蛋白質修飾
protein modification |
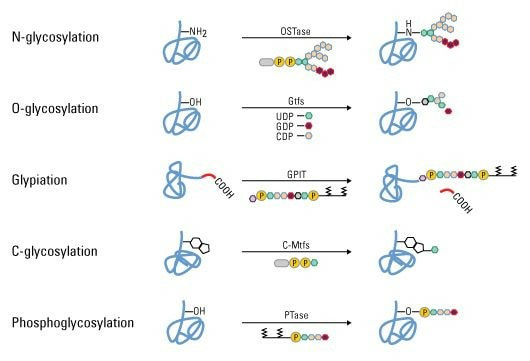
某些類型的蛋白質翻譯後修飾
Some types of protein PTMs (Post-Translational Modification ) |
https://www.creative-proteomics.com/blog/index.php/the-new-chapter-of-protein-modification-research/
The importance of protein modifications
Protein is the basic functional unit that performs cellular functions, and its expression is regulated by genomics and epigenetics. Proteins usually need to be modified to varying degrees to achieve the desired function after expression. The post-translational modification process is tightly regulated by a series of modified enzymes and de-modification enzymes, which allows the protein to exhibit a certain stable or dynamic specific function at a certain instant. Protein post-translational modification (PTM) increases the functional diversity of the proteome by covalently adding functional groups or proteins, regulating proteolytic cleavage of the subunit or degradation of the entire protein.
These modifications include phosphorylation, glycosylation, nitrosylation, methylation, acetylation, lipidation, and proteolysis, and affect almost all aspects of normal cell biology and pathogenesis. Protein phosphorylation is by far the most common PTM and has been detected in approximately 17,500 human gene products. Post-translational modification is a key mechanism for increasing the diversity of proteome. Although the genome contains only 20,000 to 25,000 genes, the proteome is estimated to contain more than 1 million proteins. Changes in transcription and mRNA levels increase the size of the transcriptome relative to the genome, and numerous different post-translational modification increases the complexity of the proteome relative to the transcriptome and genome. Therefore, identifying and understanding PTMs is critical to the study of cell biology and disease treatment and prevention.
Progress in Protein Modification Research
Due to the heterogeneity and relative abundance of post-translationally modified proteins, research of protein post-translational modification is mainly based on existing proteomics technology systems, including electrophoresis, chromatography, mass spectrometry, and bioinformatics tools. Or the peptide segment is enriched and separated, the heterogeneity caused by the modification is eliminated, and the modification site is marked to make a difference from the theoretical mass. The difference is detected by mass spectrometry to identify the protein, and the modification site is identified by tandem mass spectrometry. PTMs are widely present in eukaryotic cell biology and are critical for the signaling and life activities of organisms, but PTMs identification is often more difficult than identification of unmodified peptides.
Protein phosphorylation is the most common and most studied modification in organisms, and tyrosine phosphorylation, especially phosphorylation of tyrosine kinase receptors, has been shown to play a key role in the induction and growth of cancer cells. A variety of small molecule inhibitors and monoclonal antibodies against different tyrosine kinase receptors have also been developed as first-line drugs for the treatment of cancer. Currently, for well-studied post-translational modifications of proteins mainly include phosphorylation, acetylation, methylation and ubiquitination.
The research methods and key technologies of phosphorylation include immunoprecipitation, flow cytometry, two-dimensional gel electrophoresis, and solid phase metal affinity chromatography. The main research methods for acetylation include identification of acetylation sites by mass spectrometry, identification of acetylation sites based on acetylated antibodies that specifically recognize acetylated lysine residues, identification of acetylation by label-based methods, modification the site and so on. The main research approaches for methylation include methylation-specific PCR, bisulfite sequencing, and high-resolution melting curve.
The research methods and key technologies of glycosylation include radiolabeling, molecular fluorescent labeling, electrophoresis, lectin labeling, antibody labeling, chemical enzyme methods and so on. The types of techniques currently applied for ubiquitin proteins are relatively monotonous. The detection of ubiquitin proteins, the localization of ubiquitin targets, and the exploration of the properties of ubiquitin proteins, these techniques must be continuously improved and modified. For example, traditional protein post-translational modification studies primarily rely on specific antibody-based immunoassays or radiolabeling techniques. These methods play an irreplaceable role in the study of cellular signaling processes mediated by post-translational modifications at a single site. However, due to some shortcomings of the above-mentioned techniques, such as high operational requirements and long preparation period of specific antibodies, it is difficult to achieve large-scale detection of post-translational modification of proteins.
Progress in Protein Modification Detection Technology
In recent years, the proteomics strategy based on liquid chromatography-mass spectrometry (LC-MS) has developed rapidly, which provides a powerful research tool for protein post-translational modification research at the system level. And mass spectrometry-based proteomics greatly gives a deeper understanding of the occurrence and dynamics of protein post-translational modification. To date, quantitative proteomics has been used primarily to study PTM regulation in cell culture models, which provides new insights into the role of abnormal PTM patterns in human disease. Mass spectrometry libraries have been widely used to accurately study the quantitative analysis of various proteins, especially in the application research of quantifying known proteins.
The new strategy of immunoaffinity preconcentration of PTM peptides with monoclonal antibodies combined with liquid chromatography-mass spectrometry (LC-MS/MS) analysis has attracted much attention in many post-translational modification studies. The basic method of PTMScan technology involves the use of 9M urea lysate to lyse diseased cell lines or patient tissue blocks (enough to extract 10 mg of protein) to obtain protein lysates, which are obtained by digestion with endonuclease (usually using trypsin). Peptide mixture, the peptide mixture was purified by C18 purification column, and then the peptides containing different protein post-modification were affinity-enriched and purified by CST company’s unique protein post-translational modification motif. – Tandem mass spectrometry (LC-MS/MS) for semi-quantitative analysis.
In addition, bioinformatics analysis methods have also made progress in the field of PTM research. In recent years, with the development of proteomics technology, bioinformatics research based on proteomics, especially modified proteomics data, can provide a holistic and post-translational modification and deeper understanding. In 1999, Nikolaj Blom of the Center for Biological Sequence Analysis (CBS) used phosphorylation sites of 210 tyrosine, 584 serine and 108 threonine known at the time as a training set, and the prediction of non-specific protein phosphorylation sites was achieved for the first time by using the neural network algorithm. Since then, similar experiments have been gradually carried out, and successive studies have gradually come out. A stud which compared normal skin and skin cancer tissues in mice and combined with quantitative proteome and quantitative phosphorylation data, showed that 47.3% of proteins were found to be regulated at both protein and phosphorylation levels, while more than half of the proteins were only regulated at the regulation level. It is foreseeable that the combination of calculation and experiments, through experimental methods to verify or at least partially verify the prediction results has become a research trend in the field.
Protein is the basic functional unit that performs cellular functions, and its expression is regulated by genomics and epigenetics. Proteins usually need to be modified to varying degrees to achieve the desired function after expression. The post-translational modification process is tightly regulated by a series of modified enzymes and de-modification enzymes, which allows the protein to exhibit a certain stable or dynamic specific function at a certain instant. Protein post-translational modification (PTM) increases the functional diversity of the proteome by covalently adding functional groups or proteins, regulating proteolytic cleavage of the subunit or degradation of the entire protein.
These modifications include phosphorylation, glycosylation, nitrosylation, methylation, acetylation, lipidation, and proteolysis, and affect almost all aspects of normal cell biology and pathogenesis. Protein phosphorylation is by far the most common PTM and has been detected in approximately 17,500 human gene products. Post-translational modification is a key mechanism for increasing the diversity of proteome. Although the genome contains only 20,000 to 25,000 genes, the proteome is estimated to contain more than 1 million proteins. Changes in transcription and mRNA levels increase the size of the transcriptome relative to the genome, and numerous different post-translational modification increases the complexity of the proteome relative to the transcriptome and genome. Therefore, identifying and understanding PTMs is critical to the study of cell biology and disease treatment and prevention.
Progress in Protein Modification Research
Due to the heterogeneity and relative abundance of post-translationally modified proteins, research of protein post-translational modification is mainly based on existing proteomics technology systems, including electrophoresis, chromatography, mass spectrometry, and bioinformatics tools. Or the peptide segment is enriched and separated, the heterogeneity caused by the modification is eliminated, and the modification site is marked to make a difference from the theoretical mass. The difference is detected by mass spectrometry to identify the protein, and the modification site is identified by tandem mass spectrometry. PTMs are widely present in eukaryotic cell biology and are critical for the signaling and life activities of organisms, but PTMs identification is often more difficult than identification of unmodified peptides.
Protein phosphorylation is the most common and most studied modification in organisms, and tyrosine phosphorylation, especially phosphorylation of tyrosine kinase receptors, has been shown to play a key role in the induction and growth of cancer cells. A variety of small molecule inhibitors and monoclonal antibodies against different tyrosine kinase receptors have also been developed as first-line drugs for the treatment of cancer. Currently, for well-studied post-translational modifications of proteins mainly include phosphorylation, acetylation, methylation and ubiquitination.
The research methods and key technologies of phosphorylation include immunoprecipitation, flow cytometry, two-dimensional gel electrophoresis, and solid phase metal affinity chromatography. The main research methods for acetylation include identification of acetylation sites by mass spectrometry, identification of acetylation sites based on acetylated antibodies that specifically recognize acetylated lysine residues, identification of acetylation by label-based methods, modification the site and so on. The main research approaches for methylation include methylation-specific PCR, bisulfite sequencing, and high-resolution melting curve.
The research methods and key technologies of glycosylation include radiolabeling, molecular fluorescent labeling, electrophoresis, lectin labeling, antibody labeling, chemical enzyme methods and so on. The types of techniques currently applied for ubiquitin proteins are relatively monotonous. The detection of ubiquitin proteins, the localization of ubiquitin targets, and the exploration of the properties of ubiquitin proteins, these techniques must be continuously improved and modified. For example, traditional protein post-translational modification studies primarily rely on specific antibody-based immunoassays or radiolabeling techniques. These methods play an irreplaceable role in the study of cellular signaling processes mediated by post-translational modifications at a single site. However, due to some shortcomings of the above-mentioned techniques, such as high operational requirements and long preparation period of specific antibodies, it is difficult to achieve large-scale detection of post-translational modification of proteins.
Progress in Protein Modification Detection Technology
In recent years, the proteomics strategy based on liquid chromatography-mass spectrometry (LC-MS) has developed rapidly, which provides a powerful research tool for protein post-translational modification research at the system level. And mass spectrometry-based proteomics greatly gives a deeper understanding of the occurrence and dynamics of protein post-translational modification. To date, quantitative proteomics has been used primarily to study PTM regulation in cell culture models, which provides new insights into the role of abnormal PTM patterns in human disease. Mass spectrometry libraries have been widely used to accurately study the quantitative analysis of various proteins, especially in the application research of quantifying known proteins.
The new strategy of immunoaffinity preconcentration of PTM peptides with monoclonal antibodies combined with liquid chromatography-mass spectrometry (LC-MS/MS) analysis has attracted much attention in many post-translational modification studies. The basic method of PTMScan technology involves the use of 9M urea lysate to lyse diseased cell lines or patient tissue blocks (enough to extract 10 mg of protein) to obtain protein lysates, which are obtained by digestion with endonuclease (usually using trypsin). Peptide mixture, the peptide mixture was purified by C18 purification column, and then the peptides containing different protein post-modification were affinity-enriched and purified by CST company’s unique protein post-translational modification motif. – Tandem mass spectrometry (LC-MS/MS) for semi-quantitative analysis.
In addition, bioinformatics analysis methods have also made progress in the field of PTM research. In recent years, with the development of proteomics technology, bioinformatics research based on proteomics, especially modified proteomics data, can provide a holistic and post-translational modification and deeper understanding. In 1999, Nikolaj Blom of the Center for Biological Sequence Analysis (CBS) used phosphorylation sites of 210 tyrosine, 584 serine and 108 threonine known at the time as a training set, and the prediction of non-specific protein phosphorylation sites was achieved for the first time by using the neural network algorithm. Since then, similar experiments have been gradually carried out, and successive studies have gradually come out. A stud which compared normal skin and skin cancer tissues in mice and combined with quantitative proteome and quantitative phosphorylation data, showed that 47.3% of proteins were found to be regulated at both protein and phosphorylation levels, while more than half of the proteins were only regulated at the regulation level. It is foreseeable that the combination of calculation and experiments, through experimental methods to verify or at least partially verify the prediction results has become a research trend in the field.
■ 美國 Creative Proteomics :
世界級CRO專家。
世界上最先進的蛋白質組學平台之一。
綜合型CRO公司,全方位藥物研究開發服務
提供包括分子生物學,生物化學,系統生物學,有機化學,基因組學,生物信息學,結構生物學,臨床前和臨床研究,蛋白質體學相關的研究試劑和量產服務。還為分析,生物製藥,生命科學和化學研究提供色譜、光譜、質譜分析和微生物學產品。
世界上最先進的蛋白質組學平台之一。
綜合型CRO公司,全方位藥物研究開發服務
提供包括分子生物學,生物化學,系統生物學,有機化學,基因組學,生物信息學,結構生物學,臨床前和臨床研究,蛋白質體學相關的研究試劑和量產服務。還為分析,生物製藥,生命科學和化學研究提供色譜、光譜、質譜分析和微生物學產品。
- 世界級最先進品質蛋白質組學服務,生化分析,各式儀器分析 (色譜、光譜、質譜)。
- SCFA (Short Chain Fatty Acids)短鏈脂肪酸的專家,已完成數千個SCFA樣品 (糞便,血漿,組織,細胞等)。
- GLP (Glucagon-like Peptide) 胰高血糖素樣肽分析也有提供。
- 具有相當競爭力的市場價格。
- 非常快捷的完成周期 turnaround time。
- 各大知名醫藥研究單位的好夥伴。
蛋白質組學服務 Proteomics Service
各種蛋白質組學服務,以協助您的科學研究。 |

代謝組學服務 Metabolomics Service
從發現到目標分析的各種代謝組學服務。 |
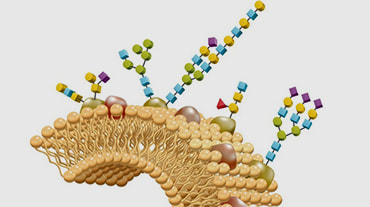
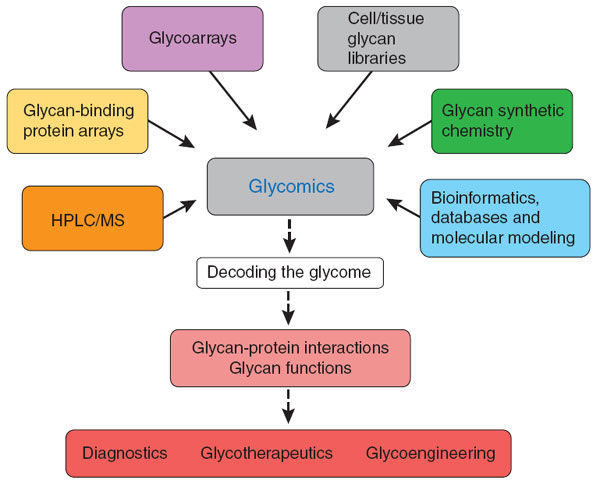
糖組學服務 Glycomics Service
幫助理解聚醣如何與特定的生物事件相關。 |
細胞分析 Cell-Based Assay
細胞分析可用於細胞遷移,粘附,侵襲和增殖。 |

生物信息學服務 Bioinformatics Service
挖掘和解釋高通量組學研究中的巨大數據。 |
客制化合成肽/蛋白質 Customized Synthesized Peptide/Proteins
為客戶提供各種合成方法和質量保證文件。 |

Customers of Creative Dynamics Inc
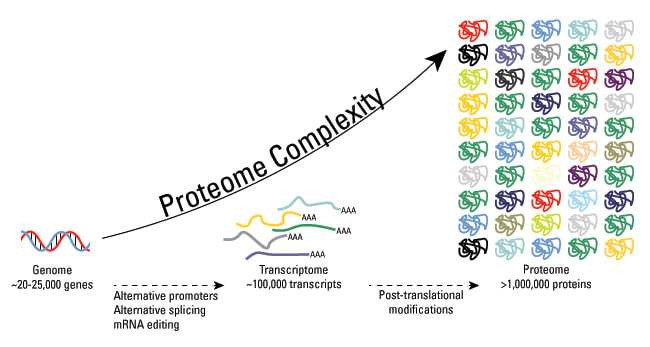
蛋白質翻譯修飾研究
Post-translational modification (PTM) PTM的表徵,包括修飾類別和修飾位點,對於細胞生物學和疾病診斷和預防的研究是至關重要的。 |
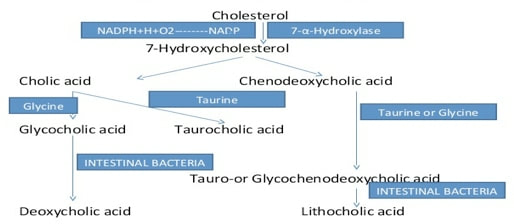
膽汁酸分析服務
Bile Acids Analysis Service 膽汁酸是一大類類固醇,在側鏈上具有羧基。 我們開發了一種靈敏,快速的方法,通過LC-MS分析66種膽汁酸樣品。 |
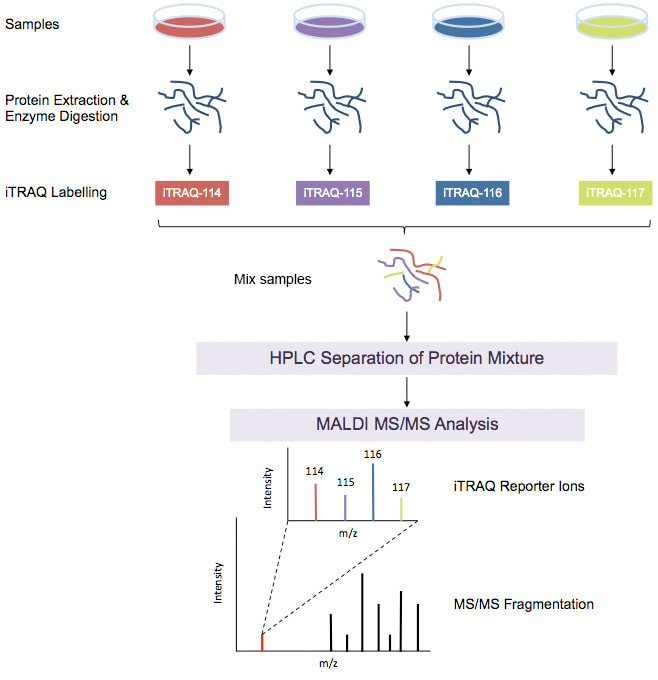
多重同量異位標籤的蛋白質組學分析
iTRAQ-based Proteomics Analysis iTRAQ使用同量異位試劑標記肽,非常適合比較正常,患病和藥物處理的樣品,時間過程研究,生物學重複,並提供相對定量信息。 |
短鏈脂肪酸(SCFAs)分析服務
Short Chain Fatty Acids (SCFAs) Analysis Service 短鏈脂肪酸(SCFA)是大腸細菌發酵膳食纖維的最終產物。 我們建立了靈敏,可靠,準確的GC-MS方法,用於量化短鏈脂肪酸 |
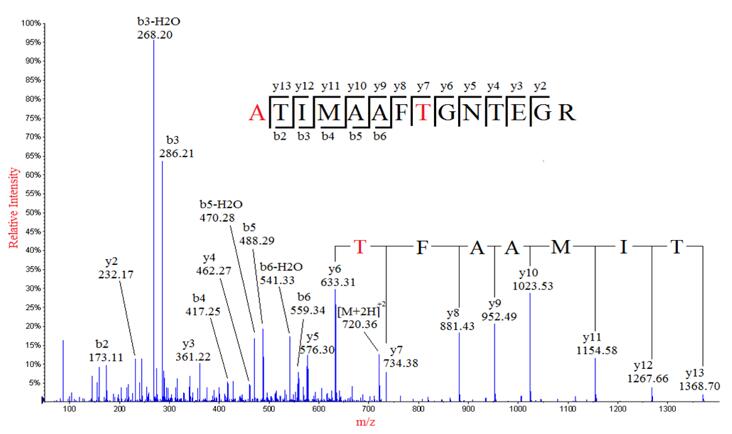
De Novo肽/蛋白質測序服務
De Novo Peptides/Proteins Sequencing Service 從頭測序是鑑定新的肽,未測序的生物體和抗體藥物的合適方法,其不能通過數據庫搜索方法檢測。 |
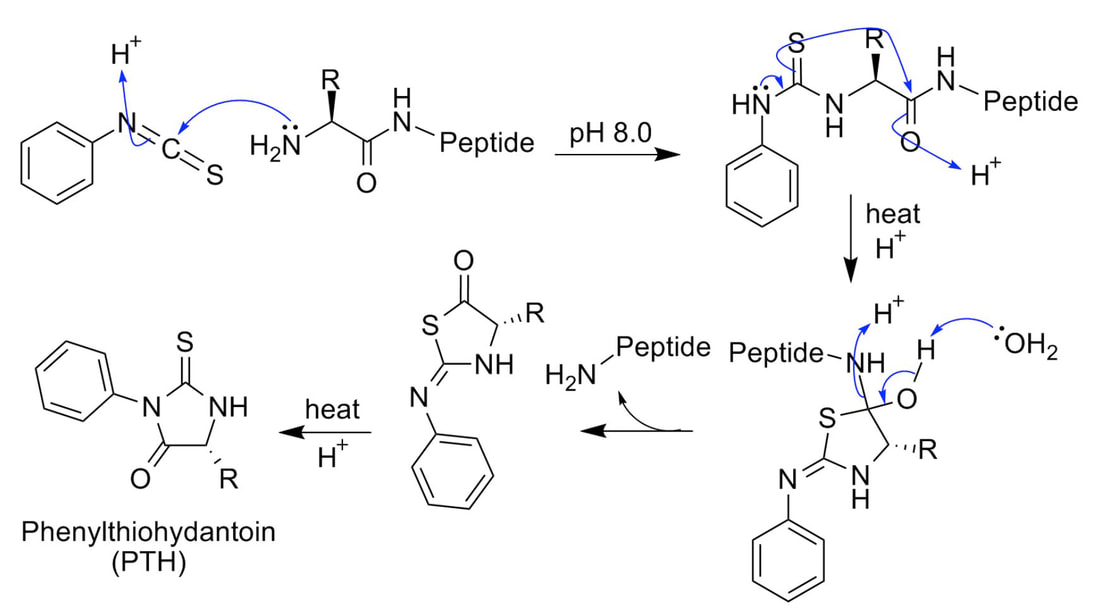
N端點Edman降階:肽或蛋白質的序列分析
N-terminal Edman degradation : Sequence Analysis of Peptides or Proteins 我們的專業蛋白質測序平台通過Edman降解或質譜(MS)提供N端序列分析,彼此具有互補優勢。 |
Creative Proteomics是Creative Dynamics Inc的蛋白質組學Proteomics部門,是家綜合CRO公司,提供包括分子生物學,生物化學,系統生物學,有機化學,基因組學,生物信息學,結構生物學,臨床前和臨床研究,制藥、生命科學研究、開發服務的全方位藥物開發服務。還為分析,生物制藥,生命科學和化學研究提供色譜、光譜、質譜分析和微生物學產品。
Creative Proteomics專門提供全方位的服務,以支持從單一蛋白質鑒定到大規模蛋白質組學研究的各種蛋白質相關研究。我們擁有世界上最先進的蛋白質組學平台之一,我們的員工科學家是經驗豐富的蛋白質組學專家。我們的蛋白質組分析平台提供蛋白質分離,特徵分析,識別鑒定和量產服務,具有高通量和超靈敏度的特點。此外,蛋白質翻譯後修飾如磷酸化和糖基化的分析是可用的。
Creative Proteomics由專業人員組成,他們在處理難以分析的樣品(包括質膜,血清,腦脊液)以及研究蛋白質翻譯後修飾和蛋白質 - 蛋白質相互作用方面具有豐富的經驗。我們的服務包括對您的項目進行全面的討論和諮詢,為您提供滿足您需求的最佳服務。與我們的合作夥伴密切合作,專業的蛋白質組學解決方案以業界最低的成本水平提供。
Creative Proteomics專門提供全方位的服務,以支持從單一蛋白質鑒定到大規模蛋白質組學研究的各種蛋白質相關研究。我們擁有世界上最先進的蛋白質組學平台之一,我們的員工科學家是經驗豐富的蛋白質組學專家。我們的蛋白質組分析平台提供蛋白質分離,特徵分析,識別鑒定和量產服務,具有高通量和超靈敏度的特點。此外,蛋白質翻譯後修飾如磷酸化和糖基化的分析是可用的。
Creative Proteomics由專業人員組成,他們在處理難以分析的樣品(包括質膜,血清,腦脊液)以及研究蛋白質翻譯後修飾和蛋白質 - 蛋白質相互作用方面具有豐富的經驗。我們的服務包括對您的項目進行全面的討論和諮詢,為您提供滿足您需求的最佳服務。與我們的合作夥伴密切合作,專業的蛋白質組學解決方案以業界最低的成本水平提供。
■ 服務項目 :
❶蛋白質組學服務 Proteomics Service
❶蛋白質組學服務 Proteomics Service
- 蛋白凝膠和成像 Protein Gel and Imaging
- 蛋白質鑒定 Protein Identification
- 蛋白質定量 Protein Quantification
- 蛋白質翻譯後修飾分析 Protein Post-translational Modification Analysis
- 自上而下蛋白質組學 Top Down Proteomics
- Peptidomics
- 蛋白質 - 蛋白質相互作用網絡 Protein-Protein Interaction Networks
- 亞細胞蛋白質組學 Subcellular Proteomics
- 其他定制實驗 Others - Customized experiments
- 非靶向代謝組學 Untargeted Metabolomics
- 靶向代謝組學 Targeted Metabolomics
- 代謝通量分析 Metabolic Flux Analysis (MFA)(MFA)
- 未知的代謝物鑒定 Unknown Metabolites Identification
- 異生代謝物分析 Xenobiotic Metabolites Analysis
- 非目標脂質組學 Untargeted Lipidomics
- 靶向脂質組學 Targeted Lipidomics
- 脂肪酸分析服務 Fatty Acids Analysis Service
- 脂肪酸衍生物分析服務 Fatty Acids Derivatives Analysis Service
- 脂肪酸代謝分析服務 Fatty Acids Metabolism Analysis Service
- 甘油脂分析服務 Glycerolipids Analysis Service
- 甘油磷脂分析服務 Glycerophospholipids Analysis Service
- 鞘脂類 Sphingolipids
- 異戊二烯 Isoprenoids
- 固醇 Sterols
- 其他分析服務 Other Analysis Services
- 外來體液體組織 Exosome LIpidomics
- MALDI成像脂質組學 MALDI-Imaging Lipidomics
- N-糖基分析 N-Glycan Profiling
- O-多醣分析 O-Glycan Profiling
- N-糖基化位點職業 N-Glycosylation Site Occupation
- O-糖基化位點職業 O-Glycosylation Site Occupation
- N-糖鏈接分析 N-Glycan Linkage Analysis
- O-糖鏈接分析 O-Glycan Linkage Analysis
- 聚醣的結構表徵 Structural Characterization of Glycans
- 糖肽分析 Glycopeptides Analysis
- 肽純度分析 Peptide Purity Analysis
- 結構活性關係(SAR)分析 Structure Activity Relationship (SAR) Analysis
- 細胞遷移分析 Cell Migration Assay
- 細胞粘附分析 Cell Adhesion Assay
- 細胞侵襲分析 Cell Invasion Assay
- 蛋白質組學的生物信息學 Bioinformatics for Proteomics
- 生物信息學代謝組學 Bioinformatics for Metabolomics
- 蛋白質的生物信息學 Bioinformatics for Protein
- 成分分析 Composition Analysis
- 流式細胞術 Flow Cytometry (FACS)
- 肽質量指紋圖譜 Peptide Mass Fingerprinting(PMF)
- De Novo肽/蛋白質測序 De Novo Peptides/Proteins Sequencing
- 肽圖 Peptide Mapping
- De Novo抗體測序 De Novo Antibody Sequencing
- 氨基酸分析(AAA)Amino Acid Analysis
- 宿主細胞蛋白質分析 Host Cell Protein Analysis
- DNA甲基化的生物分析 Bioanalysis of DNA Methylations
- 殘留的DNA測試 Residual DNA Testing
- 艾姆斯測試 Ames Test
- 細菌菌株的鑒定 Identification of Bacterial Strains

蛋白質組學服務
Proteomics Service |

代謝組學服務
Metabolomics Service |
糖組學服務
Glycomics Service |
生物信息服務
Bioinformatics Services |
細胞粘附分析
Cell Adhesion Assay |
細胞入侵檢測
Cell Invasion Assay |

細胞增殖測定
Cell Proliferation Assay |
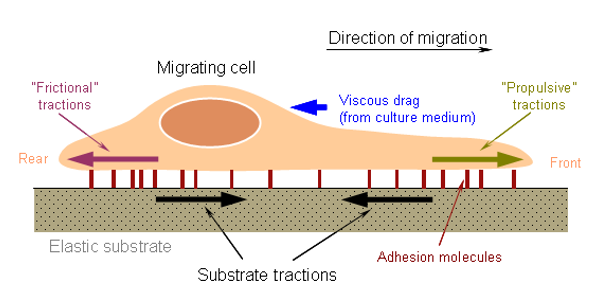
細胞遷移分析
Cell Migration Assay |
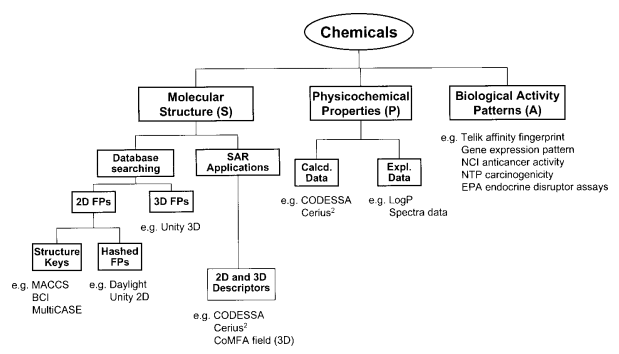
結構活動關係(SAR)分析
Structure Activity Relationship (SAR) Analysis |
■ 生化分析服務 :
❶蛋白質治療學分析 Protein Therapeutics Analysis
❶蛋白質治療學分析 Protein Therapeutics Analysis
- 蛋白質生物相似物的表徵 Characterization of Protein Biosimilar
- 蛋白質糖化分析 Analysis of Protein Glycation
- 蛋白質/肽質量的測定 Determination of Protein/Peptide Mass
- 基於MS / MS的肽測序 MS/MS based Sequencing of Peptides
- 硫醇和二硫鍵的分析 Analysis of Thiol and Disulfide Bonds
- LC-MS MRM定量 LC-MS MRM Quantification
- 翻譯後修飾分析 Analysis of Post-translational Modifications
- 蛋白質聚集分析 Protein Aggregation Analysis
- 唾液酸鑒定和定量 Sialic Acid Identification and Quantitation
- 蛋白質生物相似物的表徵 Characterization of Protein Biosimilar
- 生物仿製藥與蛋白質療法的比較 Comparison of Biosimilars and Protein Therapeutics
- 生物分子相互作用分析 Biomolecular Interaction Analysis
- N-聚醣分析 N-Glycan Analysis
- 等電點和電荷變化的測定 Determination of pI and Charge Variation
- 治療性蛋白脫肽胺的表徵 Characterization of Therapeutical Protein Deamidation
- 治療性蛋白質氧化的表徵 Characterization of Therapeutical Protein Oxidation
- 蛋白質周轉率的測定 Determination of Protein Turnover Rate
- 抗體 - 藥物偶聯物的表徵 Characterization of Antibody-Drug Conjugates
- N端截斷的分析 Analysis of N-terminal Truncation
- C端賴氨酸變體的分析 Analysis of C-terminal Lysine Variants
- N-末端焦谷氨酸的分析 Analysis of N-terminal Pyroglutamate
- 蛋白質糖化分析 Analysis of Protein Glycation
- 蛋白質PEG化的表徵 Characterization of Protein PEGylation
- 小分子的生物分析 Bioanalysis of Small Molecules
- 蛋白質的生物分析 Bioanalysis of Proteins
- ADME&PK
- 藥物研發臨床前試驗 Preclinical Trials in Drug R & D
- 生物胺分析服務 Biogenic Amine Analysis Service
- 有機酸分析服務 Organic Acid Analysis Service
- 植物激素分析服務 Plant Hormones Analysis Service
- 不飽和脂肪酸分析服務 Unsaturated Fatty Acids Analysis Service
- 碳水化合物代謝分析服務 Carbohydrate Metabolism Analysis Service
- 動物激素分析服務 Animal Hormones Analysis Service
- 食品和飲料分析 Food and Beverage Analysis
- 環境分析服務 Environment Analysis Service
- 取證分析服務 Forensics Analysis Service
- 臨床分析服務 Clinical Analysis Service
- 寵物食品分析 Pet Food Analysis
- 輔肽分析服務 Coenzyme I Analysis Service
- 生物胺分析服務 Biogenic Amine Analysis Service
- 花青素分析服務 Anthocyanins Analysis Service
- 有機酸分析服務 Organic Acid Analysis Service
- 動物激素分析服務 Animal Hormones Analysis Service
- 不飽和脂肪酸分析服務 Unsaturated Fatty Acids Analysis Service
- 植物激素分析服務 Plant Hormones Analysis Service
- 碳水化合物代謝分析服務 Carbohydrate Metabolism Analysis Service
- 三磷酸腺肽分析服務 Adenosine Triphosphate Analysis Service
- NADP分析服務 NADP Analysis Service
- 信號分子分析服務 Signaling Molecule Analysis Service
- 中草藥成分分析服務 Chinese Herbal Medicine Ingredient Analysis Service
蛋白質治療分析
Protein Therapeutics Analysis |

生物分析服務
Bioanalysis Services |


其他分析服務
Other Analytical Services |
■ 產品項目 :
❶定制的合成肽/蛋白質 Customized Synthesized Peptide/Proteins
❷穩定同位素標記全長MS蛋白質標準品 Stable Isotope Labeled Full Length MS Protein Standard
❸穩定同位素標記MS肽標準品 Stable Isotope Labeled MS Peptide Standard
❹穩定同位素標記片段蛋白質(MS標準)Stable Isotope Labeled Fragment Protein (MS Standard)
❺穩定的同位素標記分析標準 Stable Isotope Labeled Analytical Standard
❶定制的合成肽/蛋白質 Customized Synthesized Peptide/Proteins
❷穩定同位素標記全長MS蛋白質標準品 Stable Isotope Labeled Full Length MS Protein Standard
❸穩定同位素標記MS肽標準品 Stable Isotope Labeled MS Peptide Standard
❹穩定同位素標記片段蛋白質(MS標準)Stable Isotope Labeled Fragment Protein (MS Standard)
❺穩定的同位素標記分析標準 Stable Isotope Labeled Analytical Standard

定制的合成肽/蛋白質
Customized Synthesized Peptide/Proteins |
■ 儀器分析服務 :
❶色譜技術 Chromatography Technology
❶色譜技術 Chromatography Technology
- GC的分析服務 GC Based Analysis Services
- LC的分析服務 LC Based Analysis Services
- HPLC的分析服務 HPLC Based Analysis Service
- UPLC的分析服務 UPLC Based Analysis Service
- GPC的分析服務 GPC Based Analysis Service
- GPC / SEC的分析服務 GPC/SEC Based Analysis Service
- GPC-NMR的分析服務 GPC-NMR Based analysis Service
- IC的分析服務 IC Based Analysis Service
- HPTLC的分析服務 HPTLC Based Analysis Service
- DLS 分析服務 DLS Based Analysis Service
- 圓二色性的分析服務 Circular Dichroism Based Analysis Service
- FT IR 分析服務 FT IR Based Analysis Service
- NMR 分析服務 NMR Based Analysis Service
- 熒光光譜的分析服務 Fluorescence Spectroscopy Based Analysis Service
- DSC 分析服務 DSC Based Analysis Service
- DMA 分析服務 DMA Based Analysis Service
- DTA 分析服務 DTA based Analysis Service
- 三重四極桿質譜技術 Triple Quadrupole MS
- 四極桿 - 捕集器質譜技術 Quadrupole-Trap MS
- MALDI-TOF 質譜技術 MALDI-TOF MS
- LTQ Orbitrap 質譜技術 LTQ Orbitrap MS
- Q Exactive 質譜技術 Q Exactive MS
- 質譜成像 Mass Spectrometry Imaging

色譜技術分析
Chromatography Technology |
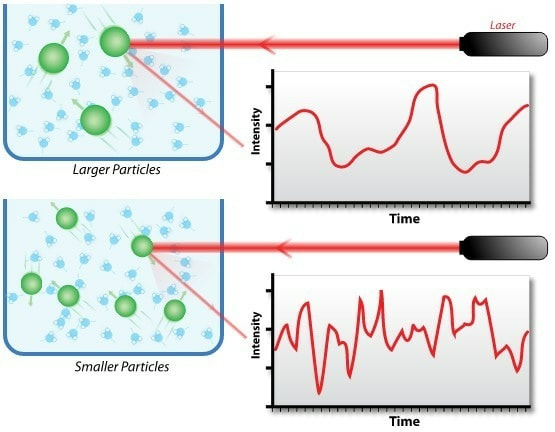
光譜技術分析
Spectroscopy Technology |
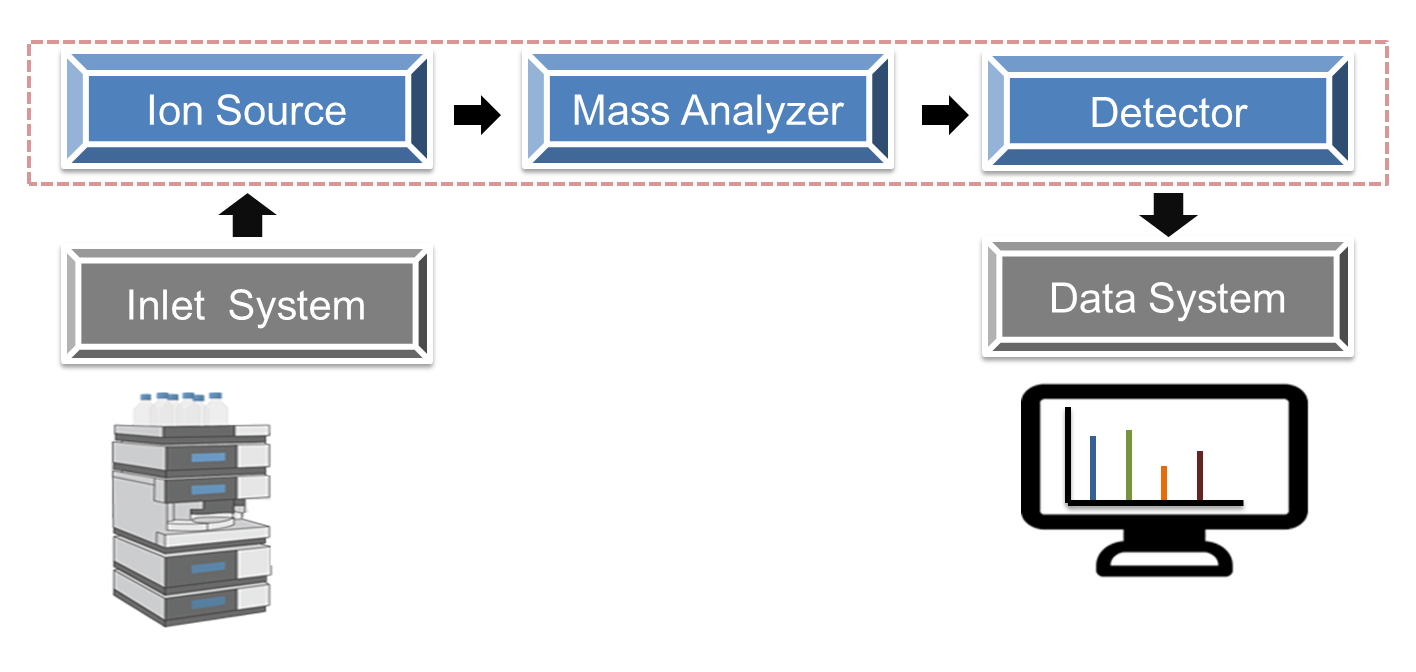
質譜儀分析
Mass Spectrometer |
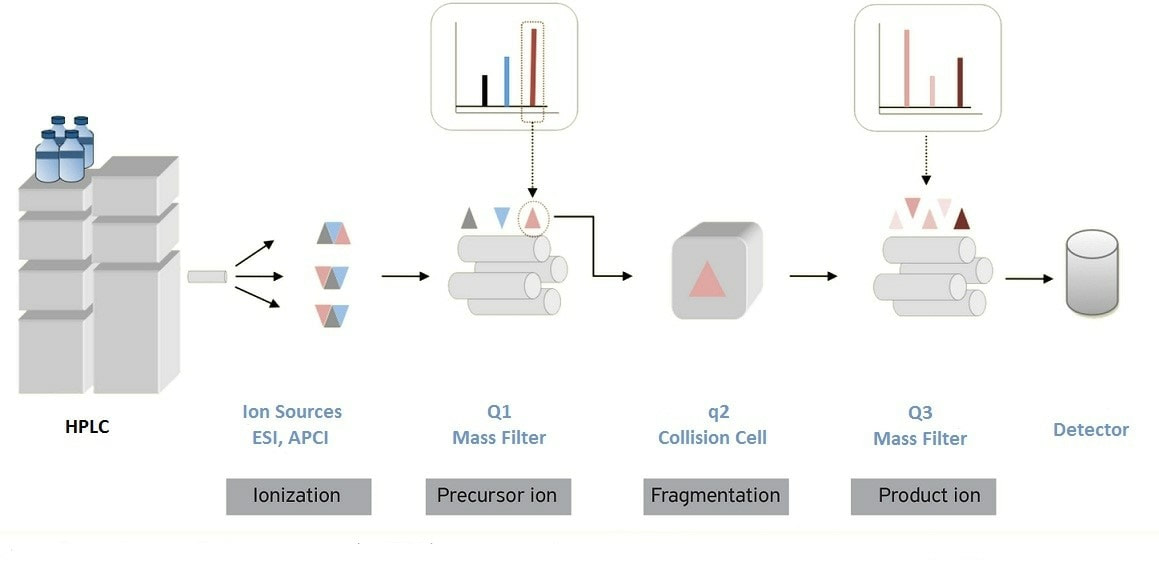
先進質譜儀分析
Innovative Mass Spectrometer |

多質譜儀分析
Mass Spectrometry Instruments |
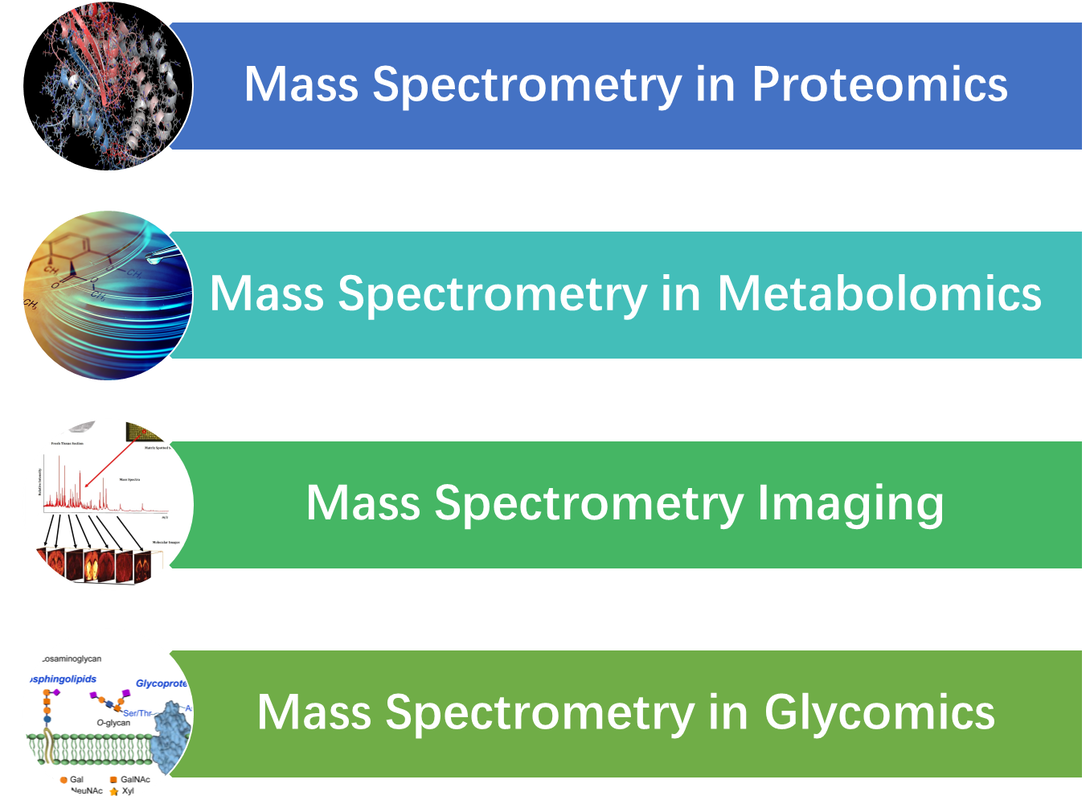
多質譜儀分析應用
Application of Mass Spectrometry |

質譜成像
Mass Spectrometry Imaging |
|
Introduction of Creative Proteomics
|

Contact E-mail : [email protected]
Phone : +886 (0)-3-5824192, +886 (0)-915-669-072 (LINE, WeChat, WhatsApp)
Copyright 2014 of EDRAGON TECHNOLOGY CORPORATION
Phone : +886 (0)-3-5824192, +886 (0)-915-669-072 (LINE, WeChat, WhatsApp)
Copyright 2014 of EDRAGON TECHNOLOGY CORPORATION
